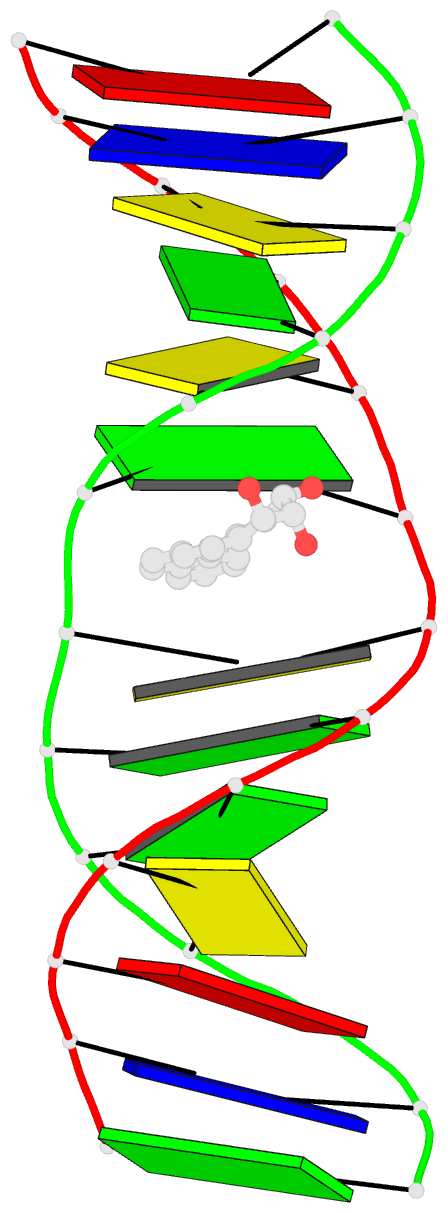
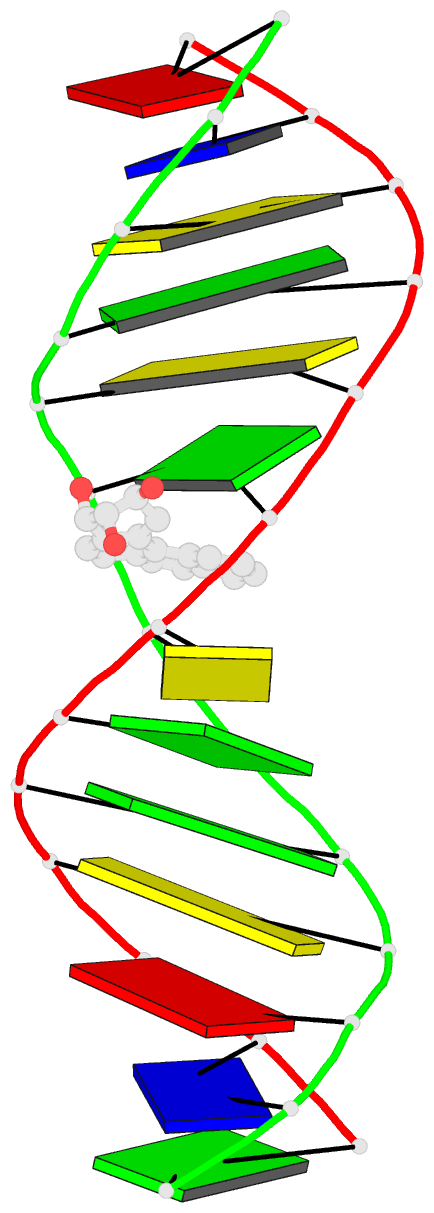
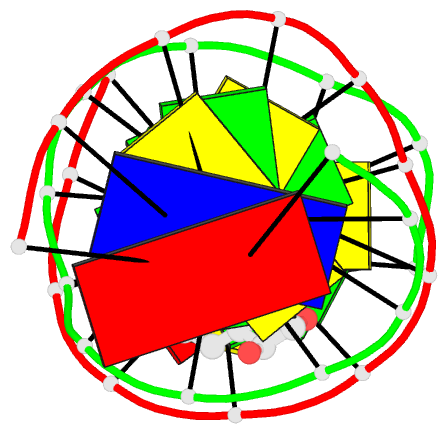
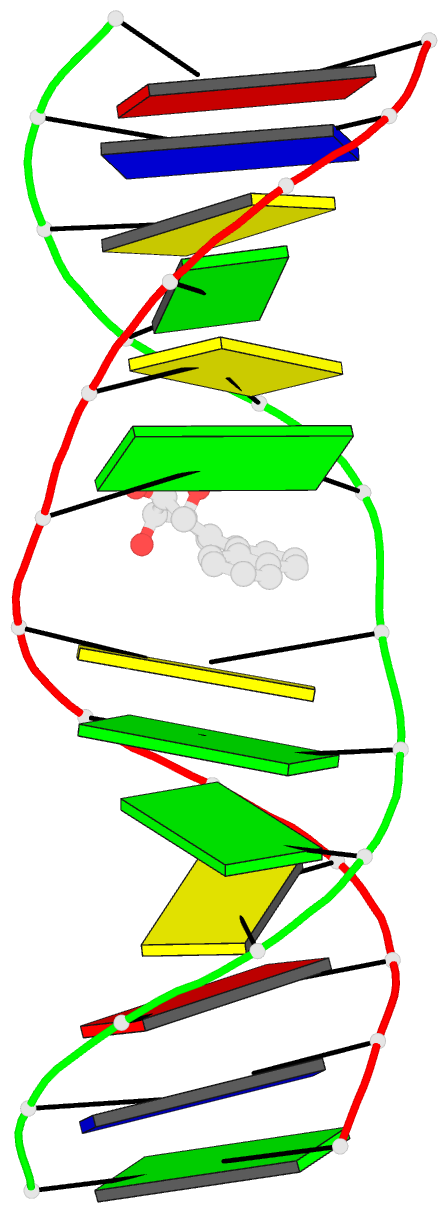
Summary information and primary citation
- PDB-id
- 2rou; DSSR-derived features in text and JSON formats
- Class
- DNA
- Method
- NMR
- Summary
- Stereospecific conformations of n2-dg 1r-trans-anti-benzo[c]phenanthrene DNA adducts: 3'-intercalation of the 1r adduct and 5'-minor groove orientation of the 1s adduct in an iterated (cg)3 repeat
- Reference
- Wang Y, Schnetz-Boutaud NC, Kroth H, Yagi H, Sayer JM, Kumar S, Jerina DM, Stone MP (2008): "3'-Intercalation of a N2-dG 1R-trans-anti-benzo[c]phenanthrene DNA adduct in an iterated (CG)3 repeat." Chem.Res.Toxicol., 21, 1348-1358. doi: 10.1021/tx7004103.
- Abstract
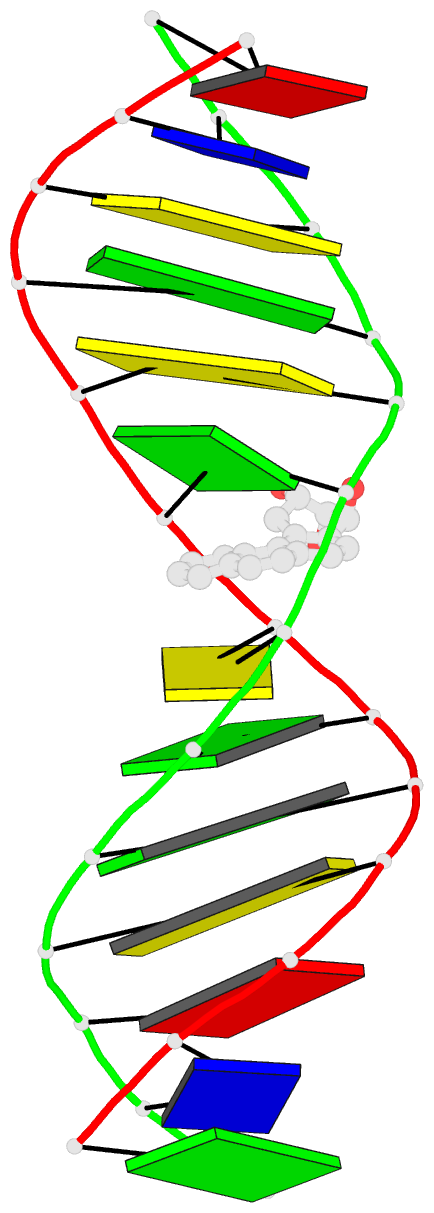
- The conformation of the 1 R,2 S,3 R,4 S-benzo[ c]phenanthrene- N (2)-dG adduct, arising from trans opening of the (+)-1 S,2 R,3 R,4 S- anti-benzo[ c]phenanthrene diol epoxide, was examined in 5'- d(ATCGC XCGGCATG)-3'.5'-d(CATGCCG CGCGAT)-3', where X = 1 R,2 S,3 R,4 S-B[ c]P- N (2)-dG. This duplex, derived from the hisD3052 frameshift tester strain of Salmonella typhimurium, contains a (CG) 3 iterated repeat, a hotspot for frameshift mutagenesis. NMR experiments showed a disconnection in sequential NOE connectivity between X (4) and C (5), and in the complementary strand, they showed another disconnection between G (18) and C (19). In the imino region of the (1)H NMR spectrum, a resonance was observed at the adducted base pair X (4) x C (19). The X (4) N1H and G (18) N1H resonances shifted upfield as compared to the other guanine imino proton resonances. NOEs were observed between X (4) N1H and C (19) N (4)H and between C (5) N (4)H and G (18) N1H, indicating that base pairs X (4) x C (19) and C (5) x G (18) maintained Watson-Crick hydrogen bonding. No NOE connectivity was observed between X (4) and G (18) in the imino region of the spectrum. Chemical shift perturbations of greater than 0.1 ppm were localized at nucleotides X (4) and C (5) in the modified strand and G (18) and C (19) in the complementary strand. A total of 13 NOEs between the protons of the 1 R-B[ c]Ph moiety and the DNA were observed between B[ c]Ph and major groove aromatic or amine protons at base pairs X (4) x C (19) and 3'-neighbor C (5) x G (18). Structural refinement was achieved using molecular dynamics calculations restrained by interproton distances and torsion angle restraints obtained from NMR data. The B[ c]Ph moiety intercalated on the 3'-face of the X (4) x C (19) base pair such that the terminal ring of 1 R-B[ c]Ph threaded the duplex and faced into the major groove. The torsion angle alpha' [X (4)]-N3-C2-N2-B[ c]Ph]-C1 was calculated to be -177 degrees, maintaining an orientation in which the X (4) exocyclic amine remained in plane with the purine. The torsion angle beta' [X (4)]-C2-N2-[B[ c]Ph]-C1-C2 was calculated to be 75 degrees. This value governed the 3'-orientation of the B[ c]Ph moiety with respect to X (4). The helical rise between base pairs X (4) x C (19) and C (5) x G (18) increased and resulted in unwinding of the right-handed helix. The aromatic rings of the B[ c]Ph moiety were below the Watson-Crick hydrogen-bonding face of the modified base pair X (4) x C (19). The B[c]Ph moiety was stacked above nucleotide G (18), in the complementary strand.