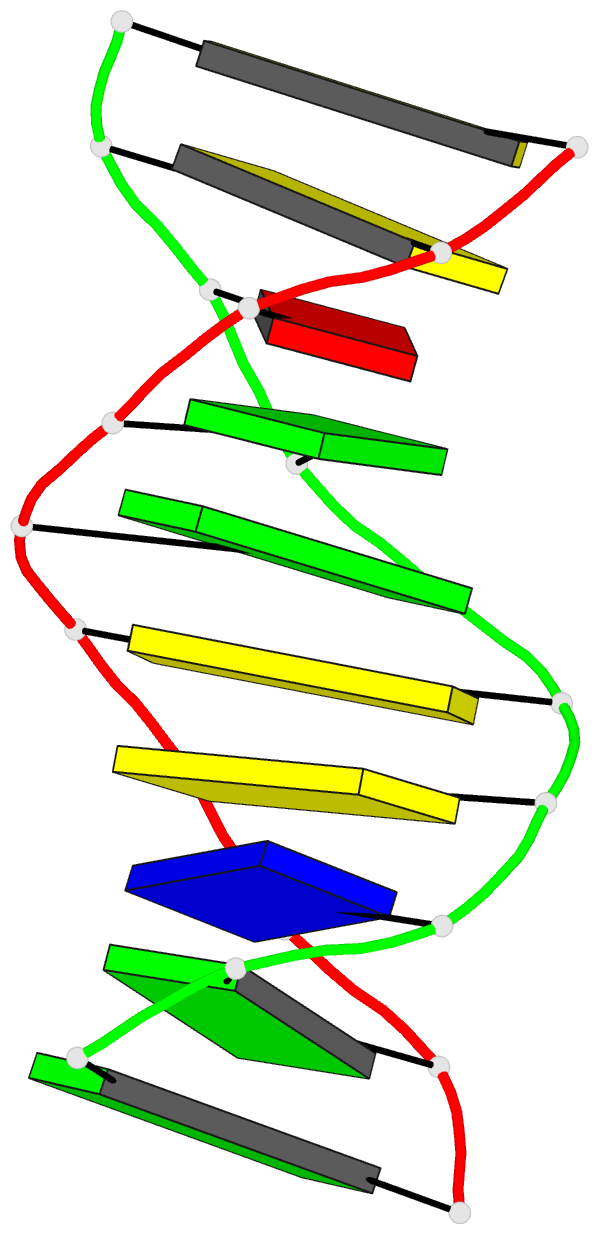
Summary information and primary citation
- PDB-id
-
122d;
SNAP-derived features in text and
JSON formats
- Class
- DNA
- Method
- X-ray (1.7 Å)
- Summary
- DNA helix structure and refinement algorithm:
comparison of models for d(ccaggcm==5==ctgg) derived from
nuclsq, tnt, and x-plor
- Reference
-
Hahn M, Heinemann U (1993): "DNA helix
structure and refinement algorithm: comparison of models
for d(CCAGGCm5CTGG) derived from NUCLSQ, TNT and
X-PLOR." Acta Crystallogr.,Sect.D,
49, 468-477. doi: 10.1107/S0907444993004858.
- Abstract
- In an earlier study [Heinemann & Hahn (1992). J. Biol.
Chem. 267, 7332-7341], the crystal structure of the
double-stranded B-DNA decamer d(CCAGGCm(5)CTGG) was refined
with NUCLSQ to R = 17.4% against 3799 2sigma structure
amplitudes in the resolution range 8-1.7 A. This structure
has now been re-refined against the same diffraction data
using either TNT or X-PLOR in order to determine to what
extent the resulting DNA conformations would differ and to
examine the suitability of these programs for the
refinement of oligonucleotide structures. The R value from
the NUCLSQ refinement could not be reached with either TNT
or X-PLOR, although both programs yield reasonably refined
DNA models showing root-mean-square deviations against the
NUCLSQ model of the decamer duplex of 0.25 and 0.32 A,
respectively. Some derived local structure parameters
differ depending on the refinement procedure used. This
holds true for several exocyclic torsion angles of the
sugar-phosphate backbone, whereas sugar puckers as well as
helical and base-pair stacking parameters are only weakly
influenced. A subset of 15 solvent sites with low
temperature factors is conserved in all three models.